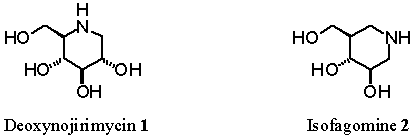Sugars in the synthesis of natural
products
PASZKÓWKA, 8-13 June 2005
ABSTRACTS
MOLECULAR SURGERY
ON PERBENZYLATED SACCHARIDES AND
CYCLODEXTRINES
Pierre SINAŸ
Ecole Normale Supérieure, Département de Chimie, UMR
CNRS 8642, 24 rue Lhomond,
75231 Paris Cedex 05, France ; e-mail :
pierre.sinaÿ@ens.fr
This lecture will present the recent extensions
of a reaction which was originally discovered in our laboratory,1
that is the regioselective de-O-benzylation of benzylated saccharides mediated
either by diisobutylaluminium hydride (DIBAL-H) or triisobutylaluminium
(TRIBAL). A remarkable development is dealing with selective deprotections on
the primary rim of perbenzylated cyclodextrins.
Such a chemistry relies both on the overall
flowerpot shape of the molecule,2 and also, in a rather unique
manner, on its directional structure.3,4
1.
M.
Sollogoub, S.K. Das, J.-M. Mallet, P. Sinaÿ, C.R. Acad. Sci. Paris Ser IIc, 1999, 441-448.
2.
A.J.
Pearce, P. Sinaÿ, Angew. Chem. Int.
Ed. 2000, 39, 1507-1508.
3.
T.
Lecourt, A.J. Pearce, A. Herault, M. Sollogoub, P. Sinaÿ, Chem. Eur. J. 2004, 12, 2960-2971.
4.
O.
Bistri, M. Sollogoub, P. Sinaÿ, unpublished results.
HIGHLY DIASTEREOSELECTIVE
SYNTHESIS OF AZASUGARS
STARTING FROM α-AMINO ALDEHYDES
Janusz JURCZAK
Department of Chemistry, University of Warsaw, 02-093 Warsaw
Institute of Organic Chemistry, Polish Academy of Sciences, 01-224
Warsaw
jurczak@icho.edu.pl
Azasugars
have generated a great deal of interest due to their ability of mimic carbohydrates
in a variety of biological processes. The known methods for the synthesis of
azasugars are mainly based on transformations of naturally occurring pentoses
and hexoses, but they can also be synthesized from nonsugar precursors. Among
them, α-amino
aldehydes are very convenient, versatile, and effective chirons.1
In this contribution, we would like to report
total syntheses of four representative azasugars, starting from suitably
protected α-amino
aldehydes, derived from L-phenylalanine, L-tyrosine, and D-serine.

- Jurczak,
J.; Gołębiowski, A. Chem. Rev. 1989, 89, 149; Reetz, M. T. Chem.
Rev. 1999, 99, 1121.
KETENE
DITHIOACETALS AS VERSATILE INTERMEDIATES FOR ONE CARBON HOMOLOGATION OF
CARBOHYDRATES: SYNTHESIS OF 3-DEOXY-2-ULOSONIC ACIDS
Jacek
MŁYNARSKI, Anna BANASZEK
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka
44/52, 01-224 Warsaw, Poland
The ketene dithioacetal formation is generally regarded
as highly efficient method for the direct one carbon homologation of ketones
and aldehydes. Typical ketene dithioacetal formation proceeds via
Horner-Emmons or Peterson olefination reaction. These compounds are extremely
useful synthetic equivalents of carbonyl derivatives as they are easily
convertible into aldehydes, carboxylic acids and esters.

As a part of long-term project on bioactive
carbohydrates we ventured into the synthesis and biological evaluation of 3-deoxy
ulosonic acids.1 This communication is focused on the application of
ketene dithioacetal methodology to the synthesis of such carbohydrates. Special
attention is paid to the unprecedented, successful formation of ketene
dithioacetals B from sugar 2-deoxy-1,5-hexonolactones A,
developed in our laboratory. Besides, these intermediates enabled the
construction of a variety isomeric 3-deoxy-2-ulosonic acids and their 2-deoxy
counterparts, among them DAH and KDO. Further, ketene dithioacetal were employed
in the direct, efficient and stereospecific synthesis of biologically relevant
KDO disaccharides.
1.
(a) Mlynarski,
J.; Banaszek, A. Trends in Organic Chemistry, 2003, 10,
51-60; (b) Mlynarski, J. and Banaszek A. Organic
Lett. 1999 1 1709-1711; (c) Mlynarski, J. and Banaszek A. Tetrahedron: Asymmetry 2000
11 3737-3746.
VINYLOXY-ALKOXIDES
-
NEW POWERFUL REAGENTS
FOR
STEREOSELECTIVE C-C BOND FORMATION -
Jessica
RICHTER
Organisch-Chemisches
Institut, Westfalische Wilhelms-Universitat,
Corrensstraße
40, 48149 Munster, Germany
Vinyloxy-ethanol
1 and derivatives are qualified reagents for carbon-carbon-coupling
reactions [1,2]. The deprotonation of 1 yields in the
corresponding vinyloxy-alkoxide. By its treatment with lewis acids intermediates
of type 2 are formed. Their reaction with carbonyl components 3 results in b-hydroxy-1,3-dioxolanes of type 4.

The
stereochemical behavior of this new type of c-c bond formation - in a formal sense
comparable to the aldol reaction - can be quite simply controlled by variation
of all variables of the system. The stereochemical information can be
introduced either by the carbonyl component A, the vinyloxy-alcohol
(substituted at the olefinic part B or in the dioxolane moiety C),
the metal reagent D or any combination of these.

By
changing one or more of these influencing factors highly diastereoselective and
enantio-selective results can be obtained[3,4,5].
References
[1] M. Schmeichel,
H. Redlich, Synthesis 1996, 1002.
[2] P. Maier,
H. Redlich, Synlett 2000, 257.
[3] P. Maier,
Ph. D. Thesis, Universität Münster 2003.
[4] D.
Vortmeyer, Ph. D. Thesis, Universität Munster 2004.
[5] P. Maier,
H. Redlich, J. Richter, D. Vortmeyer, E.-U. Würthwein, submitted.
CONVENIENT
SYNTHESIS OF COMPLEX OLIGOSACCHARIDES BY USE OF TRANS-SIALIDASE
J. THIEM,
B. NEUBACHER
Institute
of Organic Chemistry, University of Hamburg,
Martin-Luther-King-Platz
6, D-20146 Hamburg, Germany
In Chagas
disease trans-sialidase from Trypanosoma cruzi effects the transfer of
Neu5Ac from a human host cell to the cell surface of the pathogen. This unusual
transfer mechanism enables the pathogen to protect its own cell surface against
recognition of the mammalian immune system. Whereas this enzyme belongs to the
superfamily of the sialidases it shows only transferase activity if a suitable
acceptor molecule is available. Thus, trans-sialidase catalyses the
transglycosylation of several natural and
non natural Neu5Ac glycosides to Galb-R
derivatives.
In this work with
pNP-Neu5Ac as standard donor glycoside transfer could be achieved to several
different acceptor substrates leading to biologically active compounds such as the
T-antigen. Further, non naturally occuring oligosaccharides could be obtained
and subsequently used as building blocks for convenient syntheses of more
complex glycoconjugates in convincing yields.
The distinct
transferase activity and the high acceptor specificity, which excludes
monosaccharides as acceptor substrate, allows efficient aproaches to complex
oligosaccharides such as Neu5Aca2-3Galb1-4GlcNHAcaAll in a
tandem one pot synthesis. In a first step GlcNHAcaAll was
glycosylated with pNP-Gal employing b-galactosidase
from Bacillus circulans. Subsequent addition of pNP-Neu5Ac gave the
disaccharide which was in situ transsialylated with recombinant
trans-sialidase (T. cruzi).

Further, potential
donor substrates were synhesized with modifications of the Neu5Ac C7-C9
glycerol chain by single or double periodate cleaveage of the pNP-Neu5Ac
glycoside followed by reduction of the corresponding carbonyl compounds with
cyanoborohydrate. These novel Neu5Ac mimitics could be obtained in excellent
yields. Surprisingly, these unusual octunolosonic and heptulosonic acid
derivatives were recognized by trans-sialidase and transglycosylated in
comparable yields with lactose derivatives as acceptor substrates, to
accomplish a tandem one pot synthesis towards novel Neu5AcLacNAc glycoside
mimetics.
DESIGN
OF A SCAFFOLD BASED ON CARBOHYDRATES: AN APPROACH TO NATURAL PRODUCT MIMICS
A. PEDREGOSA, A. M. GÓMEZ, J. Cristóbal LÓPEZ, S.
VALVERDE
Instituto
de Química Orgánica General (CSIC, Madrid, SPAIN)
Natural products constitute one of
the main avenues for the discovery of new pharmacological interesting leads. In
this contest, there is an increasing interest in the use of polyfunctional
molecules (usually named “scaffolds”) for the preparation of mimics of natural
products with biological activity.
We
have developed in our laboratory an epoxy-exo-glycal that could be used as a
molecular scaffold. The reactivities of the three main functional groups
present in the scaffold: the exo-glycal, the vinyl epoxide and the epoxide
groups will be examined, describing the preparation of the various derivatives.
Finally, we
refer to the preparation of inhibitors of the main autolysine presente in the
pneumococcus (LytA) that could eventually suppress the virulence of these
bacterias.
APPLICATION OF METALOSALEN
COMPLEXES TO ASYMMETRIC CATALYSIS UNDER HIGH-PRESSURE CONDITIONS
Piotr
KWIATKOWSKI,a Janusz JURCZAKa,b,*
aInstitute of
Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224
Warszawa, POLAND;
bDepartment of
Chemistry, Warsaw University, Pasteura 1, 02-093Warszawa, POLAND
jurczak@icho.edu.pl
Readily available chiral metalosalen complexes
are very attractive potential catalysts, however they are not effective in many
reactions under normal conditions owing to relatively low Lewis acidity. In
some cases the solution of this problem can be application of high-pressure
technique.1
In this communication we present examples of
enantioselective reactions of simple aldehydes, catalyzed by salen chromium and
cobalt complexes under high-pressure conditions (ca.10 kbar). We
succeeded in allylation,2 hetero-Diels-Alder3 and
Friedel-Crafts reactions4 (Scheme) to obtain the desired products
with moderate to good ee’s. These chiral products are well known as convenient
precursors, particularly in syntheses of modified carbohydrates and some other
natural products having a pyran moiety.

1. High Pressure Chemistry; Eldik,
R., Klarner, F.-G., Eds.; Wiley: New York, 2002.
2. Kwiatkowski P., Jurczak J. Synlett 2005,
227.
3. (a) Malinowska M., Kwiatkowski P., Jurczak J. Tetrahedron
Lett. 2004, 45, 7693.
(b) Kwiatkowski
P., Asztemborska M., Jurczak J. Tetrahedron: Asymmetry 2004, 15,
3189.
4. Kwiatkowski P., Wojaczynska
E., Jurczak J. Tetrahedron: Asymmetry 2003, 14, 3643.
SYNTHESIS
OF LIQUID- AND SOLID-PHASE CATALYSTS FOR ENANTIOSELECTIVE TRANSFORMATIONS BASED
ON CARBOHYDRATES
Christine HOBEN, Christian BECKER, Horst KUNZ
Institut
fuer Organische Chemie, Universitaet Mainz, Duesbergweg 10-14,
D-55128 Mainz, Germany
In
general, the backbone of an asymmetric organocatalyst should be multifunctional
so it can carry several coordinating side-chains. It should be conformationally
stable and readily available. These conditions are fully met by carbohydrates
so they are predestined to replace known chiral turn elements. Besides their
high density of chiral information, they offer functional groups in abundance
to manipulate the catalysts performance by introducing additional
stereodifferentiating groups or to tie it to a polymer support.

A library of organocatalysts, based on known
systems, but with carbohydrate
backbone (1), have been synthesized and successfully employed in
enantioselective Strecker- (2) and Mannich- reactions (3),
as well as in the synthesis of cyanohydrins.
TOWARDS
MACROCYCLIC SUCROSE DERIVATIVES WITH
C2 – SYMMETRY
Sławomir JAROSZ, Arkadiusz
LISTKOWSKI
Institute of Organic Chemistry, Polish Academy of Sciences,
Kasprzaka 44/52, 01-224 Warsaw, Poland
One of the programs in our laboratory deals
with modification of sucrose molecule at the terminal positions. As a part of
this program several macrocyclic receptors containing sucrose unit were
prepared.1-3 Due to poor properties of such compounds in molecular
recognition4 we have focused our attention on preparing such
derivatives possessing C2 symmetry. Key-compound - 6’-O-tert-butyl-diphenylsilyl-1’,2,
3,’3’,4,4’-hexa-O-benzylsucrose (2), obtained from the known diol
(1)1,3 in 60% yield, was converted into 6’-O-acroyl-6-O-allyl-
(3) or 6‑[(2-p-toluenesulfonyloxy)ethyl]- (4)
derivatives. Reaction of 3 under RCM conditions gave a mixture of
products, from which the C2 symmetrical compound 5 was
isolated by HPLC as well as both internal ring-closing and one linear dimeric
products. Treatment of (4) with sodium hydride in DMF surprisingly2
afforded mostly cyclization product 6, however small amounts of the
expected compound 7 were also isolated.

- M. Mach, S. Jarosz, A. Listkowski, J. Carbohydr.
Chem., 2001 (20) 485-493;
- S. Jarosz, A. Listkowski, M. Mach, Polish J. Chem., 2001
(75) 638-687;
- S. Jarosz, A. Listkowski, J. Carbohydr. Chem.,
2003 (22) 753-763;
- S. Jarosz, A. Listkowski, B. Lewandowski, Z.
Ciunik, A. Brzuszkiewicz, Tetrahedron, 2005, accepted.
THE
LIAISON BETWEEN HYPERVALENT IODINE REAGENTS AND CARBOHYDRATE CHEMISTRY
Andreas KIRSCHNING
Institute of Organic Chemistry, Universiy
of Hannover, Schneiderberg 1B, 30167 Hannover, Germany
Hypervalent (III) reagents are known since 1886 when Willgerodt described
dichloro iodosobenzene for the first time. Only during the past two decades
hypervalent iodine reagents in the oxidation states +1 to +5 have seen a broad
interest among synthetic organic chemists.1 Besides the Dess-Martin
periodinane, IBX has recently appeared as a versatile oxidation agent on the laboratory shelves. Hypervalent
iodine reagents in the oxidation state +3 show resemblance to organometallic
reagents as they undergo ligand exchange reactions as well as reductive
eliminations. Particularly the latter property allows to perform unique
oxidations which rarely has been exploited in natural product synthesis including
carbohydrate chemistry.
Over the
past decade we have developed several synthetic applications for saccharides
using iodine(III) reagents. The report
will focus on these new synthetic applications.2 These will include
oxidations, glycosidations and other transformations. In addition, mechanistic
studies and applications in solid-phase assisted synthesis will be addressed.
_____________
1 A. Varvoglis, Hypervalent
Iodine in Organic Synthesis, Academic Press, San Diego 1997
M. Ochiai, Top. Curr. Chem. 2003, 224,
5-68.
2 A. Kirschning, Eur. J. Org. Chem. 1998, 2276-2274.
BIODIVERSITY
EXPLOITATION FOR THE SEARCH OF BIOACTIVE SUGARS
Amélia Pilar RAUTER
Departamento
de Química e Bioquímica, Faculdade de Ciências da Universidade de Lisboa,
Edifício C8, 5º Piso, 1749-016 Lisboa, Portugal.
E-mail:
aprauter@fc.ul.pt; Tel: +351 217500952; Fax: +351 217500088
Bioactive natural products present a wide
variety of chemical structures, being some structural units responsible for a
great diversity of bioactivities, namely lactones, heterocyclic rings and
flavonoid moieties. In this work we will report on the development of synthetic
strategies leading to sugar derivatives which contain those bioactive units,
among others. The evaluation of their biological activities will also be
presented and the results obtained will be discussed and correlated to their
structure and stereochemistry.
Chain elongation, followed by the construction
of lactones or heterocyclic moieties linked to position 4 of the furanose ring,
gave compounds exhibiting neuroactivity in insects, presenting some of them a
remarkable insecticidal activity against flies. One of the pseudo-C-nucleosides
inhibited butyrylcholinesterase, an enzyme involved in neurotransmission in the
brain. Its inhibition has been found to exert a beneficial therapeutic effect
in some patients suffering from Alzheimer’s disease.
Efficient and direct approaches to macrocyclic
bislactones or five-membered ring lactones linked/fused to furanose/pyranoside
moieties will be described and discussed their structure/fungicidal efficacy
relationship.
Trichloroacetimidates and glycals were the glycosyl
donors investigated for the preparation of flavonoid O-glycosides. Among
them, the synthesis of anthocyanidin glycosides starting from
trichloroacetimidates was particularly challenging, considering the properties
of these pigments related to their solubility and instability. The experimental
procedure was easy to carry out, leading to the stereoselective synthesis of
the target molecules in moderate yield.
Glycals led to the stereoselective synthesis of 2-deoxy-O-glycosides
a-anomer or to the corresponding
Ferrier products, when promoted by triphenylphosphine hydrobromide or acid
zeolite, respectively. These reactions were extended to other acceptor
molecules such as sterols, sugars, thiols and heterocyclic bases. When
aliphatic alcohols were the glycosyl acceptors, some of the 2-O-glycosides
obtained exhibited surface active and antibacterial properties, being one of
them selective over Bacillus species including Bacillus cereus, a
human pathogen bacteria close related to Bacillus anthracis.
RHODIUM-CATALYZED INTRAMOLECULAR CONJUGATE
ADDITION OF VINYLSTANNANES TO 2,3-DIHYDRO-4-PYRIDONES. AN EFFICIENT ROUTE TO
STEREOSELECTIVE CONSTRUCTION OF AZABICYCLIC RING SYSTEMS
Bartłomiej FURMAN
Institute of Organic Chemistry, Polish Academy of Sciences, 01-224
Warsaw, Poland
Indolizidine
and quinolizidine skeletons can be found in many important natural products.
These nitrogen derivatives occur in plants, insects and amphibians and exhibit
notable biological activities. Therefore, the stereoselective synthesis of
these bicyclic skeletons has become an important goal for synthetic chemists in
the recent year.
In
connection with our interest in the synthesis of azabicyclic ring systems herein,
we report a general and highly stereoselective approach to the construction of
indolizidine and quinolizidine ring skeletons, based on intramolecular
conjugate addition of vinylstannanes to 2,3-dihydro-4-pyridones catalyzed by
rhodium(I)-complex.

The experimental details as well as scope and
limitation of this novel cyclocondensation will be reported.
ENZYMATIC
SIALYLATION OF Galb1-3GalNAc DERIVATIVES
LEADING TO BIOACTIVE STRUCTURES
Agnes SCUDLO, Lars KRÖGER, Björn NEUBACHER, Joachim
THIEM
Institut für Organische Chemie, Universität Hamburg
Martin-Luther-King-Platz
6
20146
Hamburg, Germany
The sialyloligosaccharide Neu5Acα2-3Galβ1-3GalNAc
and a range of corresponding motives play an important role in Nature. They are
found in Lewis type I structures and Thomsen - Friedenreich antigen (sialyl-T
antigen) occurring in higher animals, viruses, bacteria, protozoa and
pathogenic fungi [1]. There is considerable interest in such structures with
functionalities significant for glyco-pharmaceuticals, and these studies
contribute to alternative ways for more facile preparations.
Starting from galactose and
N-acetyl-galactosamine Galβ1-3GalNAc structures could be obtained. They could be sialylated in α2-3 and α2-6 position using the
transglycolytic activity of the sialidases from C. perfringens and S. typhimurium [2]. Further enzymatic
syntheses could be achieved with a recombinant trans-sialidase from T. cruzi.

[1] A. Varki, Glycobiology, 3 (1993) 97-130
[2] D. Schmidt, B. Sauerbrei, J. Thiem,
J. Org. Chem., 65
(2000) 8518-8526
SYNTHESIS OF SUGAR-DERIVED N-VINYL OXAZOLIDINE-2-THIONES AS TEMPLATES
FOR STEREOCONTROLLED CYCLOADDITIONS
Sébastien TARDY,a Arnaud TATIBOUET,a Gilles DUJARDIN,b Patrick ROLLINa
a Institut de Chimie Organique et Analytique
– UMR 6005, F-45067 Orléans-Cedex 2, France
b UCO2M - UMR 6011, Université du Maine, F-72085 Le Mans-Cedex 9, France
sebastien.tardy@univ-orleans.fr
The synthesis and evaluation of
cyclic thionocarbamates grafted onto carbohydrate scaffolds catch a lot of
attention in our laboratory.
The nucleophilic reactivity of the
nitrogen-site in 1,3-oxazolidine-2-thiones is significant and allows N-vinylsulfonylation
through Michael addition on 1,2-bis-(phenylsulfonyl)ethylene (BPSE). The
related N-vinyl derivatives are obtained by reductive desulfonylation.1
Such N-vinyl oxazolidine-2-thiones constitute a new class of dienophilic substrates well-suited for various types of
stereocontrolled cycloadditions.
Within a joint project with UMR 6011,2 a range of
sugar-derived N-vinyl oxazolidine-2-thiones was tested as
2p
component in a [4+2] inverse electron demand hetero-Diels-Alder reaction as
shown below :
Gaulon,
C. ; Dhal, R. ; Chapin,
T ; Maisonneuve, V. ; Dujardin, G. J. Org. Chem., 2004,
4192-4002.
DESIGN, SYNTHESIS AND
BIOLOGICAL EVALUATION OF
SUGAR-DERIVED RAS
INHIBITORS
F. PERI, C. AIROLDI, E. MARTEGANI, S.
COLOMBO, F. NICOTRA
University of
Milano-Bicocca
Department of Biotechnology and Bioscience
P.za della Scienza, 2; 20126 Milano, Italy
cristina.airoldi@unimib.it
The pharmacological modulation of mutated,
tumorigenic RAS proteins activity could represent an efficient strategy to
prevent tumour formation and development. Oncogenic versions of RAS are present
in about 30% of human tumours and contain point mutations which cause the
constitutive protein arrest in its active state.
Our purpose is to develop small molecules able
to bind RAS preventing the nucleotide exchange GDP/GTP required for the protein
activation.
A class of compounds presenting an analogous
activity was described by the Schering-Plough Research Institute1.
These inhibitors are nevertheless chemically unstable and poorly water soluble2.
In the light of these evidences, we decided to transfer their putative
pharmacophore groups on a conformational rigid bicyclic scaffold derived from
the natural sugar D-arabinose (see fig. 1), in order to obtain a suitable
pharmacophores orientation for binding with RAS and to increase the water
solubility of our new compounds.

figure 1
In this
communication, the chemical synthesis and the biological activity both in
vitro and in vivo of these new
RAS inhibitors are presented.
1.
Taveras, A. G. et al. Bioorganic
and Medicinal Chemistry, 1997, 5, 125-133.
2.
a) F. Peri. et al. The
Italian Journal of Biochemistry Special Issue: SIB-BIB 2003, 2003, 52,
31.
b) S. Colombo, F. Peri, R. Tisi, F. Nicotra, E. Martegani, Ann NY Acad Sci 2004, 1030, 52-61
THE FUNCTION OF
CARBOHYDRATES IN FULLY SYNTHETIC GLYCOPEPTIDE ANTITUMOUR VACCINES
Horst KUNZ, Stefanie KEIL, Sebastian DZIADEK,
Sven WITTROCK, Constanze BROCKE
Institut fuer Organische
Chemie, Universitaet Mainz, Duesbergweg 10-14, D-55218 Mainz, Germany
Immunological differentiation of
normal cells and tumour cells needs to identify cell surface structures selectively
occurring on tumour tissues and to direct the immune response towards these
target structures. Using allylic anchors, acid-sensitive anchors or recently
developed 2-phenyl-2-(trimethylsilyl)ethyl linkers,1) glycopeptides
with the structure of tumour-associated saccharide antigens and peptide
sequences of the tandem repeat region of the polymorphic epithelial mucin MUC 1
have been synthesised on solid-phase.
The required glycosyl
amino acid building blocks 1 and 2 carrying the tumour-associated Sialyl-Tn-
and Sialyl-T antigen sidechains are generated by chemical and chemoenzymatic
methods and then applied to the solid-phase synthesis of the target
glycopeptides.2)

Based on these glycopeptide syntheses complex
constructs combining tumour-associated antigens and T cell epitopes have been
obtained which induce the proliferation of cytotoxic (CD8-positive) T cells.3) Glycopeptides with full tandem repeat sequences of MUC 1 and MUC 4 have
been synthesised and are presently investigated in terms of their conformation
and immunological properties.4,5)
1) M. Wagner, H. Kunz, Angew. Chem. 114, 315 (2002); Z.
Naturforsch. 57b, 928
(2002);
M. Wagner, S. Dziadek, H. Kunz, Chemistry
Eur. J., 9, 6018 (2003).
2) Recent review: H. Herzner, T. Reipen, M.
Schultz, H. Kunz, Chem. Rev. 100, 4495 (2000).
3) S.
Keil, C. Claus, W. Dippold, H. Kunz, Angew.
Chem. Int. Ed. 40, 366 (2001).
4)
C.
Brocke, H. Kunz, Synthesis 2004, 525.
5)
S. Dziadek, C. Brocke, H. Kunz, Chemistry Eur. J., 10,
4150 (2004).
AN ENTRY TO OXYGEN ANALOGS OF
PENICILLIN AND CEPHALOSPORIN
Marek
CHMIELEWSKI
Institute of Organic Chemistry
of the Polish Academy of Sciences, 01-224 Warsaw, Kasprzaka 44/52, Poland
The present contribution focuses
attention on the problem of stereocontrol in the formation of a desired
configuration of the bridgehead carbon atom in the title compounds. Two
synthetic methods leading to the basic skeletons of clavams and 5-oxacephams
are discussed. One involves cycloaddition reaction between vinyl ethers or
alkoxyallenes and isocyanates The second one involves the nucleophilic
substitution at C-4 of the azetidin-2-ones performed as intramolecular process.
The first method seems to be most advantageous since it allows syntheses not
only oxacephams but also clavams 1-4 related to natural clavams (5,6).
So far attempts to use the second methodology for clavams formation were
unsuccessful.

[2+2]Cycloaddition
of chlorosulfonyl isocyanate to chiral alkoxyallenes proceeds with a moderate
stereoselectivity providing, after the intramolecular alkylation of the β-lactam nitrogen atom, cephams
having an exo-propylidene group. Particulary attractive are
alkoxyallenes derived from 1,3-benzylidene L-erythritol since [2+2]cycloadducts
create an entry to the 3,4-disubstituted-5-oxacephams suitable for the
introduction of substituents not only at C-7 carbon atom, but also introduction
of a carboxylic function at the C-2 (Scheme). The last transformation has been
demonstrated using 3-keto-5-oxacepham derived from lactic acid providing
desired compound in low yield only.
Scheme

SYNTHESIS
OF MACROCYCLIC RECEPTORS CONTAINING SUCROSE SCAFFOLD
Sławomir JAROSZ, Bartosz LEWANDOWSKI, Arkadiusz LISTKOWSKI
Institute
of Organic Chemistry Polish Academy of Sciences
ul. Kasprzaka 44/52 01-224
Warsaw, bartlew81@o2.pl
 Starting from sucrose as a substrate,
we have developed an efficient method for the preparation of
1’,2,3,3’,4,4’-hexa-O-benzylsucrose (1).1 Reaction of 1 with a series of poliethylene
glycol ditosylates afforded a wide variety of chiral crown ether analogues.1,2,3
Starting from sucrose as a substrate,
we have developed an efficient method for the preparation of
1’,2,3,3’,4,4’-hexa-O-benzylsucrose (1).1 Reaction of 1 with a series of poliethylene
glycol ditosylates afforded a wide variety of chiral crown ether analogues.1,2,3
 We then focused our interest on the
synthesis of crown ether analogues containing nitrogen atoms in the macrocyclic
ring. Compound 2 has already been obtained2 and the synthesis
of other nitrogen containing receptors is still under investigation.
We then focused our interest on the
synthesis of crown ether analogues containing nitrogen atoms in the macrocyclic
ring. Compound 2 has already been obtained2 and the synthesis
of other nitrogen containing receptors is still under investigation.
Association constants of the synthesized
macrocycles with Li+, Na+ K+, NH4+
have been determined on the basis of the NMR titration experiments.
The macrocycles obtained (2-5; R = Bn) were tested as catalysts in enantio-selective addition
of carbo anions to chalcone, however, with
little success (max ee = 22%).3
1. microreview: Jarosz,
S.; Mach, M. Eur. J. Org. Chem., 2002, 769 – 780.
2. Jarosz, S.; Listkowski, A. J. Carbohydr. Chem., 2003, 22, 753 – 763
3. Jarosz, S; Listkowski, A.; Lewandowski, B.; Ciunik, Z.;
Brzuszkiewicz, A., Tetrahedron 2005, accepted
NOVEL
OLIGOAMINOGLYCOSIDES
Thomas JÖGE, Andreas KIRSCHNING*
*Institute of Organic
Chemistry, Schneiderberg 1b, 30167 Hannover, Germany.
Changing the
conformation of nucleic acids at will would enable man to interfere with the
life cycle of cells and viruses. RNA with its huge conformational diverse space
(e.g. TAR RNA of HIV‑1) is a very promising target for such an approach.
Aminoglycosides like Kanamycin A 1 are prominent for their good binding
properties to RNA.
 1
1
This
project focuses on the chemical and biological behavior of novel aminosugars. In
this context, our efforts are governed by the goal to design novel “artificial“
aminoglycosides or disaccarides like 4. These novel structures consist
of aminated sugar building blocks which are connected to each other by a
flexible linker.

Their
oligomeric character containing several amino groups is essential for efficient
binding and should lead to cooperative effects and hence tighter binding. Their
synthesis is achieved by metathesis reactions starting from allyl linker
building blocks like 3. This synthetic strategy yields
extended aminosugar structures like 4 in a few steps1-3.
1 A. Kirschning, G.-w. Chen, Tetrahedron Lett. 1999, 40,
4665-4668.
2 A. Kirschning, G.-w. Chen, Chem. Eur. J. 2002, 8,
2717-2729.
3 A. Kirschning, M. Lindner, Tetrahedron 2004, 60,
3505-3521.
THE STANNYL-PRINS REACTION. A NOVEL METHOD FOR THE SYNTHESIS OF
DIHYDROPYRANS
Magdalena DZIEDZIC
Institute of
Organic Chemistry, Polish Academy of Sciences, 01-224 Warsaw, Poland
Tetrahydropyranes are structural
features of a variety of biologically active natural products such as
poliethers antibiotics, marine toxins and pheromones. The literature now
contains many versatile methods for the synthesis of substituted pyranes, such
as the hetero-Diels-Alder reaction, the intramolecular Sakurai reaction and
ring-closing olefin methathesis. Unfortunatelly, the Sakurai approach involves
a lengthy synthesis of precursor, while the methathesis approach requires the
synthesis of complex precursors. By
contrast, the Prins cyclization, which involves treatment of a
homoallilic alcohol with a carbonyl compounds and usually mineral or Lewis
acids overcomes many of the drawbacks of the alternative methods.
Herein, we report the Lewis acid mediated
stannyl-Prins reaction as a rapid route to the dihydropyran skeleton. An
optimised reaction system of the Prins-type cyclization was observed using
TMSOTf as a Lewis acid in diethyl ether.
These results and their application
to the synthesis of more complex molecules will be discussed.
FROM
SUGARS TO SUGAR MIMICS:
STEREOSELECTIVE
SYNTHESIS OF AMINOCYCLOPENTANOLS AS GLYCOSIDASE INHIBITORS.
Inge LUNDT
Technical
University of Denmark
Department
of Chemistry, Building 201
DK 2800
Kgs. Lyngby, Denmark
The mechanism of enzymatic cleavage of
glycosides has been continuously under debate and design of new glycosidase
inhibitors has been based on structural similarity of putative intermediates or
transition states.
Recently the aminocyclopentanols have drawn
considerable attention as potent glycosidase inhibitors. Aminocyclopentanols
having a substitution pattern similar to common carbohydrates, and with the
amino group next to the side chain, has been considered as anomer selective
glycosidase inhibitors,1 since the configuration at the carbon
having the amino substituent might be mimicking either the a- or b-anomer of a substrate.
We have syntesised a range of aminocyclopantanols
with the general structures 3 and 4, starting from the bicyclic
cyclopentane-lactones 1 or 2, which are readily available from
bromodeoxyaldonolactones by a radical induced carbocyclisation.2 The
synthesis of the aminocyclopentanols and their inhibitory properties will be
presented.

1: (a) M. Kleban, P.
Hilgers, J. N. Greul, R. D. Kugler, J. Li, S. Picasso, P. Vogel, V. Jäger, CHEMBIOCHEM,
2001, 5, 365. (b) J.N. Greul, M. Kleban, B. Schneider, S. Picasso, V.
Jäger, CHEMBIOCHEM, 2001, 5, 368.
(b) A. Blaser, J.-L. Reymond, Helv. Chim.
Acta, 2001, 84, 2119; L. G. Dickson, E. Leroy, J.-L.
Reymond, Org. Biomol. Chem., 2004, 2, 1217.
2:
Johansen, S. K.; Lundt, I. J. Chem. Soc.,
Perkin Trans. 1, 1999
AZASUGARS AND AZASPIRONUCLEOSIDES
José FUENTES MOTA
Departamento de Química Orgánica, Facultad de Química,
Universidad de Sevilla, Apartado 553, E-41071, Sevilla,
Spain.
In recent years much effort has been
directed to the syntheses of iminocyclitols (also known as azasugars)1,
a type of structural analogue of sugars in which the ring oxygen atom is
replaced by a nitrogen atom. Some azasugars are naturally occurring compounds,
and in general are related to natural alkaloids. They have importance as glycosidase
inhibitors2, as they interfere with carbohydrate
recognizing-receptors, and consequently
are used in the therapy of diabetes, AIDS, and cancer3. Recently,
the first azasugar medicine has been launched1.
In this communication, we describe a
versatile route “The glycosylenamine-azaanhydrosugar route” to prepare five-,
six-, and seven-membered iminocyclitols (3)
starting from easily available glycosylenamines (1). The key chiral intermediates are anhydroazasugar derivatives (2).

The use of anhydroazasugars in the
preparation of azasugar thioglycosides (2-thioalkoxypiperidines) (4)
and of furanoid thioglycosides
of 5-aminosugars (5) is also
reported.

Finally, pyranoid and furanoid
spiro-N-mesylazetidines (6), a new
type of water-soluble spiro-C-nucleoside, are prepared from easily available
sugar spiroacetals.
(1) Afarinkia, K.; Bahar, A. Tetrahedron: Asymmetry 2005, 16, 1239-1287.
(2) Lillelund, W.H.; Jensen, H.H.; Liang, X.; Bols,
M. Chem. Rev. 2002, 102, 515-553.
(3) Le Merrer, Y.; Poitout, L. Depezay, J.C.;
Dosbaa, I.; Geoffroy, S. ; Foglietti, M.J. Bioorg. Med. Chem. 1997, 5, 519-533.
We thank the Junta de Andalucia
(FQM-134) and Ministerio de Ciencia y Tecnologia (BQU2001-3740 and
CTQ2004-1178) for financial support.
ENANTIOSELECTIVE ALLYLATION OF ACITVATED ALDEHYDES CATALYZED BY
(SALEN)Cr(III) COMPLEXES
Wojciech
CHAŁADAJ,a Piotr KWIATKOWSKI,a Janusz JURCZAKa,b
a Institute of Organic Chemistry,
Polish Academy of Sciences, 01-224 Warsaw, Poland
b Department of Chemistry,
University of Warsaw, 02-093 Warsaw, Poland
jurczak@icho.edu.pl
The addition of allylic organometallics to
aldehydes leads to homoallylic alcohols, compounds of particular importance in
the organic synthesis. For more than one decade, a range of enantioselective
catalytic systems, especially for allylation of simple aromatic and aliphatic
aldehydes, has been developed.1
We focused our attention on enantioselective allylation
of particular class of active aldehydes, namely 2-oxoaldehydes 1,
leading to homoallylic alcohols 2, compounds of significant importance
in the synthesis of highly oxygenated biologically active compounds, like
sugars and their derivatives.
We found that the reactions of
various 2-oxoaldehydes 1 with allylstannanes proceed smoothly when
catalyzed by a (salen)chromium(III) complex of type 3. The influence of
reaction variables, such as temperature, concentration, quantity of catalyst
and type of solvent. Additionally, we investigated the influence of the
structure of substrates and catalyst on the stereochemical reaction course.
Thus, we developed an efficient and undemanding method for allylation of
activated aldehydes 1 with satisfactory yields and enantiomeric excesses
up to 90% and 77%, respectively.2
1.
Denmark, S.E.; Fu,
J. Chem. Rev. 2003, 103, 2763
2.
Kwiatkowski, P., Chaładaj, W., Jurczak, J. Tetrahedron
Lett. 2004, 45, 5343
ANALOGS OF THE DNA-CLEAVING ANTIBIOTIC LEINAMYCIN
Ákos SZILÁGYI, Pál HERCZEGH
Department of Pharmaceutical Chemistry, University of
Debrecen and
Research Group for Chemistry of Antibiotics of the
Hungarian Academy of Sciences
H-4010 Debrecen, Hungary
A synthetic
introduction of the „warhead” of
leinamycin into nucleosides will be discussed.

Leinamycin
The
following nucleoside derivatives have been prepared from simple nucleosides.


SYNTHESIS OF GLUCOSINOLATES, CHEMICAL AND BIOLOGICAL TAGS IN BRASSICALES
Patrick ROLLIN
ICOA – UMR 6005,
Université d’Orléans, B. P. 6759, F-45067 Orléans, France
All vegetables in the
Brassicale order contain glucosinolates
(GSL) – anciently mentioned [1] and strikingly bio-relevant [2] thiosaccharidic metabolites which display a remarkable structural
homogeneity : a hydrophilic b-D-glucopyrano framework
bearing a O-sulfated anomeric (Z)-thiohydroximate moiety connected to a generally hydrophobic
aglycon side chain R. In the over 120 known
GSL, R is the sole structural variant, in which diversified aliphatic,
arylaliphatic or heterocyclic atom arrangements can be found.[3]
Present in all
GSL-containing plants, myrosinase (thioglucoside glucohydrolase EC 3.2.3.1) is
the unique enzyme able to effect hydrolytic cleavage of the anomeric C-S bond
of GSL; the detached aglycons undergo a fast Lossen rearrangement to mainly
produce in situ strongly electrophilic isothiocyanates and/or closely related
thiofunctionalized compounds.
Extraction
of GSL from vegetable sources is usually not a straightforward operation :
synthetic routes to naturally occurring GSL have therefore been developed over
the past decades,[4, 5, 6] then more recently extended to the elaboration of
tailor-made artificial GSL-like structures, with a view to exploring the
recognition process of myrosinase, estimating the relative importance of topical
zones in the active site and searching for enzyme inhibitors.[7]
A survey
of synthetic approaches to GSL will be presented.
[1]
Horatius, Satira IV 65-8 BC,
Liber secundus, verses 15-17
[2] Robiquet, P. J. J.
Pharm. 1831, 17,
279
[3]
Fahey, J. W.; Zalcmann, A. T.; Talalay, P. Phytochemistry, 2001, 56,
5-51.
[6]
Gil, V.; MacLeod, A. J. Tetrahedron 1980, 36, 779-783.
[7] Bourderioux, A.; Lefoix, M.; Gueyrard, D.;
Tatibouët, A.; Cottaz, S.; Arzt, S.; Burmeister, W. P.; Rollin, P. Org. Biomol. Chem., 2005, in press, and references therein.
SYNTHESIS OF GLYCO-AMINO ACIDS AND PEPTIDES
Hermen S. OVERKLEEFT, Gijsbert GROTENBREG, Mattie S. M.
TIMMER, Gijsbert A. VAN DER MAREL, Mark OVERHAND
Leiden Institute of Chemistry, Leiden University, P. O.
Box 9502, 2300 RA Leiden, The Netherlands
Monosaccharides
have long been recognized as versatile building blocks in synthetic organic
chemistry. They are readily available from natural sources and are
characterized by a wealth of functional, conformational and stereochemical
variations. They are widely used in natural product synthesis and in the
development of compounds with desirable biological or therapeutical properties.
Research efforts over the past decades have accumulated a wealth of
information, enabling the manipulation of each individual
functional group in a given monosaccharide
building block almost at will.
This paper presents our recent
results concerning the use of carbohydrates as cheap, chiral and enantiopure
starting materials in the construction of a variety of sugar amino acids, and
their evaluation as both carbohydrate and peptide mimetics. Further, our latest
results in the development of a novel Ugi-type three-component reaction of
sugar derived azido-aldehydes will be discussed.
Recent key publications:
- M. S. M. Timmer, M. Verdoes, L. A. J. M. Sliedregt, G. A. van der Marel, J. H. van Boom and H. S. Overkleeft, The use of a mannitol-derived fused oxacycle as a combinatorial scaffold, J. Org. Chem. 2003, 68, 9406.
- S. H. L. Verhelst, B. Paez Martinez, M. S. M. Timmer, G. Lodder, G. A. van der Marel, H. S. Overkleeft and J. H. van Boom, A short route toward chiral, polyhydroxylated indolizidines and quinolizidines, J. Org. Chem. 2003, 68, 9598.
- G. M. Grotenbreg, M. S. M. Timmer, A. L. Llamas-Saiz, M. Verdoes, G. A. van der Marel, M. J. van Raaij, H. S. Overkleeft and M. Overhand, An unusual turn structure adopted by a furanoid sugar amino acid incorporated in gramicidin S, J. Am. Chem. Soc. 2004, 126, 3444.
- G.
M. Grotenbreg, A. E. Christina, A. E. M. Buizert, G. A. van der Marel, H. S.
Overkleeft and M. Overhand, Synthesis and application of carbohydrate-derived
morpholine amino acids, J. Org. Chem.
2004, 69, 8331.
SPIRONUCLEOSIDES AND
PSEUDOSPIRONUCLEOSIDES
José M. ILLANGUA
Departamento de Química
Orgánica, Facultad de Química,
Universidad de Sevilla,
Apartado 553, E-41071, Sevilla, Spain.
The
chemistry of spironucleosides, a type of nucleoside in which the anomeric
carbon belongs simultaneously to the sugar ring and to the nitrogenated
heterocyclic moiety, has received a considerable development in the last decade
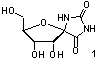
especially from the isolation of (+)-hydantocidin (1), the first natural spironucleoside,1 which shows low toxicity for mammals
and has herbicidal and plant growth-regulatory
activities. Since 1993, syntheses of (+)-hydantocidin2 and many spirofuranoid derivatives of
different heterocycles, pyranoid analogues of hydantocidin, and carbocyclic
derivatives have been reported3.
However, syntheses of pseudospironucleosides, in which the
spiranic carbon atom is C2 or C3 of the sugar ring, are very scarce4 , despite the potential interest of these
compounds as precursor of novel conformationally restricted nucleosides,
related to compounds with demonstrated anti-HIV and anti-virus activities5.
In this
communication, we report the preparation of new spironucleosides and 2- and
3-pseudospironucleosides using isothiocyanates as key intermediates. We
describe the stereocontrolled synthesis of thiohydantoin spironucleosides and N-alkyl, aryl and glycosyl derivatives
starting from isothiocyanatoulosonates or aminoulosonates (Scheme 1).
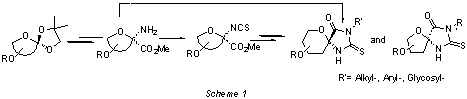
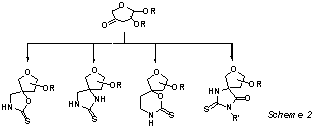
We also
describe the syntheses of 3-(and 2-) pseudospironucleosides from 3- (and 2-) uloses via an intermediate
thioureido derivative ( Scheme 2).
[1]
Haruyama, H; Takayanna, T. J. Chem. Soc. Perkin Trans. I 1991, 1637-1640.
2 Mio, S.; Ichinose,
R.; Goto, K.; Sugai, S. Tetrahedon,
1991, 47, 2111-2120. (b) Mio, S.; Kumagawa, Y.; Sugai, S.; Tetrahedron 1991, 47, 2133-2144.
(c) Matsumoto, M.; Kirihara, M.;
Yoshino, T.; Katoh, T.; Terashima, S. Tetrahedron
Lett. 1993, 34, 6289. (d) Chemla, P. Tetrahedron
Lett. 1993, 34, 7391-7394. (e) Harrington, P.; Jung, M. Tetrahedron Lett. 1994, 35, 5145.5148. (f) Nakajima, N.;
Matsumoto, M.; Kirihara, M.; Hashimoto, M.; Katoh, T.; Terashima, S. Tetrahedron, 1996, 52, 1177-1194.
3 (a) Taillefunier, C.; Thielges, S.; Chapleur, Y., Tetrahedron 2004, 60, 2213-2224. (b) Renard, A. ;
Lhomme,J. ; Kotera, M. J.Org. Chem.
2002, 67, 1302-1307.(c)Long, D. D.; Smith, M.D.; Muller, M.; Fleet,
G.W.J. J. Chem. Soc. 2002, 1982-1998, (d) Somsák, L.; Nagy,
V.; Docsa, T.; Tóth, B.; Gergely, P. Tetrahedron:
Asymmetry 2000, 11, 405-408 (e) Gasch, C.; Pradera,
M.A.; Salameh, B.A.B.; Molina, J.L.; Fuentes, J. Tetrahedron: Asymmetry 2001,
12, 1267-1277 (f) See also Freire,
R.; Martín, A.; Pérez-Martín, I.; Suárez, E. Tetrahedron Lett. 2002, 43, 5113-5116 and references cited
therein.
4
Nguyen Van Nhien, A.; Ducatel, H.; Len,C.; Postel, D. Tetrahedron Lett., 2002,
43, 3805-3808.
5 (a)
Nguyen et al. Pharmacy and Pharmacology
2001, 53, 939-943. (b) Camarasa et al. J. Med. Chem. 2005, 48, 1158-1168 and references cited therein.
SYNTHESIS AND BIOLOGICAL
EVALUATION OF SOME HIGHER HOMOLOGUES OF KNOWN POTENT IMINOSUGAR-BASED
GLYCOSIDASE INHIBITORS
Yves BLÉRIOT
Ecole Normale Supérieure, Département de Chimie, UMR
CNRS 8642, 24 rue Lhomond,
75231 Paris
Cedex 05, France ; e-mail : yves.bleriot@ens.fr
Glycosidase inhibitors have been the
subject of strong interest in the past two decades due to their therapeutic potential
in the treatment of diabetes, HIV, viral infections and cancer. The
design of glycosidase inhibitors is usually based on the mimic of the oxycarbenium-like transition
state. To this end, a great number of five and six-membered iminocyclitols have
been synthesized, where the endocyclic oxygen atom or the anomeric carbon of
the parent sugar have been replaced by a nitrogen atom such as in
deoxynojirimycin 1 and isofagomine 2 respectively.1 Despite
interesting biological properties, much less efforts have been put into the
synthesis of seven-membered iminocyclitols.2
As part of an ongoing project on new
carbohydrate mimetics, we were interested in the design of new
seven-membered iminocyclitols.3 The increased flexibility of such structures
associated with the unusual spatial distribution of the hydroxyl groups should
allow a new glycosidase inhibition profile for these molecules.
The chemical synthesis and the
inhibition on glycosidases of some higher homologues of known potent six-membered
ring iminosugar-based glycosidase inhibitors will be presented.
1.
Iminosugars as glycosidase inhibitors ; A. Stütz, Ed. ;
Wiley-VCH : Weinheim, 1999.
2.
X.-H.
Qian, F. Moris-Varas, C.-H. Wong, Bioorg.
Med. Chem. Lett. 1996, 6, 1117-1122.
3. H. Li,
Y. Blériot, C. Chantereau, J.-M. Mallet, M. Sollogoub, Y. Zhang, E. Rodriguez-
Garcia, P. Vogel, J.
Jimenez-Barbero, P. Sinaÿ, Org.
Biomol. Chem. 2004, 2, 1492-1499
ANOMERIC HYDROPEROXIDES:
SYNTHESIS, ENANTIOSELECTIVE EPOXIDATION
Wioletta
KOŚNIK, Marek CHMIELEWSKI
Institute of Organic Chemistry
Polish Academy of Sciences, Kasprzaka 44, 01-224 Warsaw, POLAND
Relatively stable hydroperoxides 1-5
have been used for enantioselective oxidation of prochiral alcohols and sulfides
in the presence of Ti(OiPr)4 with stereoselectivities varied
from about 10 to 50% e.e.1 They have, however, several significant
drawbacks such as: the accessibility, relatively lower asymmetric induction,
selfoxidation, and can not be regenerated to be used again after reoxidation
since hemiacetals obtained from them are unstable and rearrange to
α,β-unsaturated aldehydes.2 Epoxidation of electrophilic
olefins with anomeric hydroperoxides in the presence of a base, in principle,
does not remove drawbacks of the reagents mentioned above.3
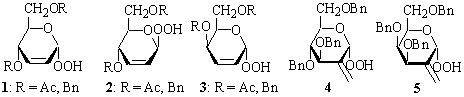
Oxidation of readily available
2-deoxysugars or their methyl glycosides with 50 % hydrogen peroxide in
dioxane in the presence of sulfuric acid3 provides corresponding
hydroperoxides 6-12 in 48-75 % yields. They are relatively stable
and can be separated into pure anomers by chromatography; compounds 8
practically exist as single anomers only.
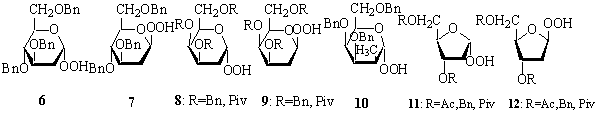
Experiments with the use of anomeric
hydroperoxides 6-12 as chiral oxidants were performed using
2-methyl-1,4-naphtoquinone (13) under standard conditions provided by
Taylor et al.3 to afford epoxyquinone with e.e in the range 28-48%.
After asymmetric epoxidation of electrophilic olefins, the hemiacetal can be
regenerated from the post reaction mixture and reoxidized again to
corresponding hydroperoxide.
1.
Chmielewski, M.; Jurczak,
J.; Maciejewski, S. Carbohydr. Res., 1987,
165, 111; Hamann, H.-J.;
Höft, E.; Chmielewski, M.; Maciejewski, S. Chirality, 1990, 5, 338; Hamann, H.-J.; Höft, E.;
Mostowicz, D.; Mishnev, A.; Urbańczyk-Lipkowska, Z.; Chmielewski, M. Tetrahedron,
1997, 53, 185;
Mostowicz, D.; M. Jurczak, M.; Hamann, H.-J.; Höft, E.; Chmielewski, M. Eur. J. Org. Chem., 1998, 2617.
2.
Fraser-Reid, B.;
Radatrus, B. J. J. Am. Chem.
Soc., 1970, 92, 5288;) Gonzales, F.; Lesage, S.;
Perlin, A. S., Carbohydr. Res., 1975, 42,
267; Torsel, K. Tyagi, M. P.; Acta Chem. Scand., 1977, B31, 297; Tatsuta, K.; Yamauchi, T.; Kinoshita, M., Bull.
Chem. Soc. Japan, 1978,
51, 3035; Chmielewski, M. Polish J. Chem., 1980, 54, 1913.
3.
Dwyer,
C.L.; Gill, Ch.D.; Ichikawa, O.; Taylor, R.J.K. Synlett, 2000,
704; Bundu, A.; Berry, N.G.; Gill,
Ch.D.; Dwyer, C.L.; Stachulski, A.; Taylor, R.J.K.; Whittall, J., Tetrahedron:
Asymmetry, 2005, 16, 283.
SYNTHESIS OF C-GLYCALS VIA DIETHYLZINC-MEDIATED UMPOLUNG OF p-ALLYL PALLADIUM DERIVED
FROM 1-EXO-METHYLENE 2,3-ANHYDROFURANOSES
A. BARRIO, A. M. GÓMEZ, J. C. LÓPEZ,
S.VALVERDE
Our group has studied the formation
of p-allyl palladium complexes (2)
derived from 1-exo-methylene 2,3-anhydrofuranoses (1).
Intermediates type (2) had already been shown to react with
nucleophiles to obtain C-glycals (3)1.
More recently, we have studied a new
synthetic approach to C-glycals (4) based on the reaction of p-allyl palladium complexes (2) with
electrophiles rather than nucleophiles. In this context, the reaction of 2
with Et2Zn results in the umpolung of the p-allyl palladium complex and allows its
coupling reaction with carbonyl compounds.
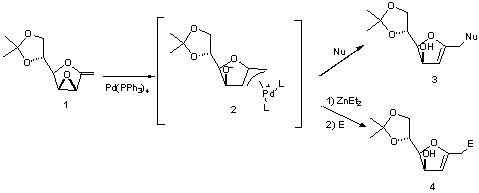
1.
Gómez, A. M.;Pedregosa, A.;Valverde, S.;López, J. C. Chem. Commun. 2002, 2022
GLYCIDIC SCAFFOLDS IN DRUG RESEARCH
Francesco NICOTRA
Dipartimento di Biotecnologie e
Bioscienze, Università degli Studi di Milano-Bicocca,
I-20126 Milano, Italy
Recent efforts in the use of
carbohydrates as original scaffolds for the production of bioactive compounds
will be reported. Orthogonally protected and solid phase supported
glycostructures have been used for the production of libraries, exploiting the
combinatorial approach by derivatisation of each hydroxyl group with different
pharmacophores. Example of peptidomimetics synthesised on a carbohydrate
skeleton properly orienting amino and carboxylic residues will be described. In
order to increase the conformational rigidity of the sugar templates, a variety
of original bicyclic or policyclic polifunctionalised structures have been
synthesised from carbohydrates. Same of them have spiro or condensed bicyclic
structures, others include one or more sugars in a macrocyclic framework or in
cyclopeptides in order to induce bioactive peptide loops. New strategies for
the synthesis of iminosugars libraries will be reported, and finally modified
Lipid A with antagonistic activities will be also described.
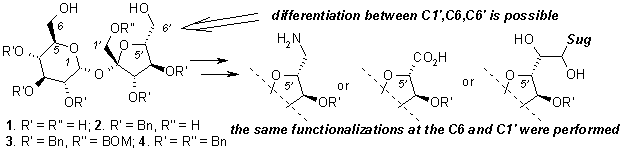
WHAT CAN BE DONE FROM SUCROSE ???
Sławomir JAROSZ
Institute of Organic Chemistry, Polish Academy of
Sciences, Kasprzaka 44/52, 01-224 Warszawa, POLAND; sljar@icho.edu.pl
Sucrose (1) is available in large
quantity on the market; its annual production exceeds 100 mln tons. Purity of
the commercially available disaccharide is so high that it may be used as
reagent for chemical transformation without any additional purifications what
makes this molecule potentially useful source of chirality for chemical
synthesis. As a part of an on-going program we have elaborated the convenient
method of the synthesis of partially protected sucrose in which all secondary
hydroxyl groups are protected as benzyl ether (2). The primary ones can be differentiated, which allows to prepare
a wide variety of sucrose analogs modified at each terminal position (C1’, C6,
C6’).1
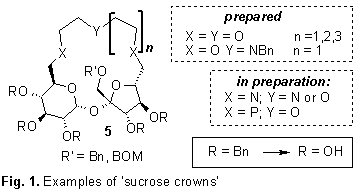 The diols with the hydroxyl groups
free at the C-6, and C6’ positions (3 and
4) were used for the preparation of the
macrocyclic crown ether analogues with incorporated sucrose unit. Selected
examples are shown on Fig. 1.
The diols with the hydroxyl groups
free at the C-6, and C6’ positions (3 and
4) were used for the preparation of the
macrocyclic crown ether analogues with incorporated sucrose unit. Selected
examples are shown on Fig. 1.
Stability constants of these
receptors with cations of the first group (Li, Na, K) and also NH4+
were measured by the NMR titration method. The macrocycles 5 (R = R’ = Bn) were also used as chiral catalysts in the Michael
addition of carboanions to chalcone with, however, little success.2
1.
microreview: Jarosz, S.; Mach, M. Eur. J. Org. Chem., 2002,
769 – 780.
2.
Jarosz,
S; Listkowski, A.; Lewandowski, B.; Ciunik, Z.; Brzuszkiewicz, A. Tetrahedron, 2005, accepted
Approaches toWARDS THE SYNTHESIS of
miharamycinS
SUGAR MOIETY
Filipa MARCELO,1 Amélia P. RAUTER,1
Yves BLÉRIOT,2 Pierre SINAŸ2
1Departamento de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa Edifício C8,
5º Piso, 1749-016 Lisboa, Portugal
2 École Normale Supérieure, Département de Chimie,
UMR 8642, 24 Rue Lhomond ,
75231 Paris Cédex 05, France
Miharamycins are complex nucleoside
antibiotics produced in low yield by Streptomyces
miharaensis. They act as potent inhibitors of Pyricularia oryzae, now considered as a bioterrorism agent, known
to cause the rice blast disease.
In this communication we would like
to report on the strategies used to build up the miharamycins bicyclic
carbohydrate moiety 1, starting from
the protected D-glucose derivatives 2 and 3.

The synthetic pathways investigated
are based on modifications of the previously described procedure starting from
compound 2 [1], as well as on a
different strategy based upon transformations of the monosaccharide 3 by regioselective oxidation,
stereoselective Wittig reaction, cis-diol
addition, cyclisation, and reduction.
[1]
Fairbanks, A. J.; Sinaÿ, P. Synlett
1995, 95, 1859-1876.
A DOUBLE ASYMMETRIC
INDUCTION IN 1,3-DIPOLAR CYCLOADDITION OF A CYCLIC NITRONES DERIVED FROM MALIC
ACID AND TARTARIC ACID WITH UNSATURATED γ-LACTONES
Sebastian
STECKO, Konrad PAŚNICZEK, Margarita JURCZAK, Marek CHMIELEWSKI
Institute of Organic Chemistry of the Polish Academy of Sciences, 01-224
Warsaw, Poland
1,3-Dipolar cycloaddition of
lactones 1 and 2 with nitrones 3 and 4 provides
adducts 5-11.1 In the case of the nitrone 3 and
lactones 1 and 2 only one adduct was formed, 5 and 8,
respectively, as a result of the exo addition, anti to t-butoxyl
at C-3 of the dipole. On the other hand, the nitrone 4 with both
lactones 1 and 2 affords corresponding exo adducts 7,
10 and 11 which are accompanied by endo ones 6 and 9.
This result should be compared with the same reactions performed on
δ-lactones. In all, so far, investigated cases, the formation of endo
adducts was not observed.
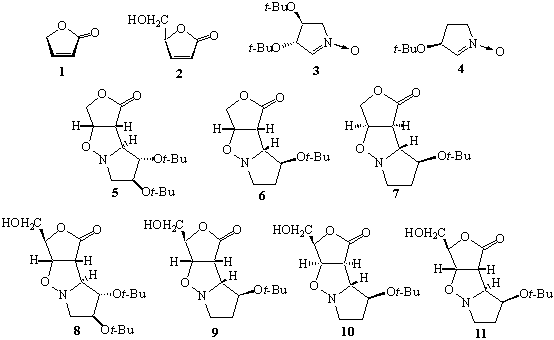
1. Pasniczek, K.; Socha, D.; Jurczak, M.;
Frelek, J.; Suszczyńska, A.; Urbańczyk-
Lipkowska, Z. Chmielewski, M. J.
Carbohydr. Chem., 22, 613 (2003).
2.
Jurczak, M.; Mostowicz, D.; Panfil, I.; Rabiczko, J.; Socha, D.;
Chmielewski, M. in
Targets in Heterocyclic Systems, Attanasi, O., Ed.; Springer: Berlin, 2001;
Vol. 5, p. 59.
HIGHLY DIASTEREOSELECTIVE
ALLYLATION
OF CHIRAL OXIME ETHERS
Joanna
CHAŁKO,a Janusz JURCZAKa,b
a Department of Chemistry, University
of Warsaw, 02-093 Warsaw
b Institute of Organic
Chemistry, Polish Academy of Sciences, 01-224 Warsaw
jurczak@icho.edu.pl
Various derivatives of allylamines
are widely used in the preparation of natural products, including
carbohydrates.1 The methods for the preparation of chiral allyl
amines and their derivatives are mainly based on the diastereoselective
nucleophilic addition of allylometallic reagents to the C=N bond.
We found that among many compounds
bearing the azomethine group, aldoxime ethers are interesting substrates for
the asymmetric allylation reaction. As a convenient substrate we have chosen O-alkyloximes
derived from glyoxylic acid modified by Oppolzer’s chiral auxiliary.
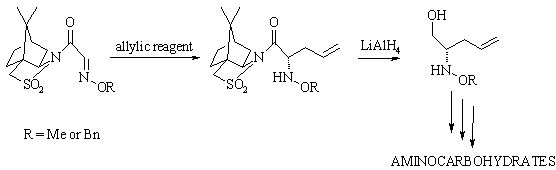
The best results were obtained when
allylation was carried out under Barbier conditions. In this case, we were able
to get desired O-alkilhydroxylamines as
a single
(2S)-enantiomer and with 55% overall yield.
1. Trost, B. M.; Van Vranken, D. L. J.
Am. Chem. Soc. 1993, 115, 444
RECENT PROGRESS IN THE
PREPARATION OF SOME GLYCOCONJUGATES AND ANALOGUES FROM EASILY AVAILABLE
CARBOHYDRATE BASED-SYNTHONS
Yves QUENEAU
Laboratoire de Chimie Organique, UMR 5181 CNRS-UCBL-INSA
INSA, Bât. J. Verne, 20 avenue A. Einstein, 69621 Villcurbanne Cedex,
France
tel +33 (0)4 72 43 61 69; fax. +33 (0)4 72 43 88 96; e-mail: yves.qucncau@insa-lyon.fr
Carbohydrate-containing natural
products such as glycopeptides, glycolipids, as well as oligosaccharides are
present about everywhere in living systems and are responsible for numerous and
important biological processes. There is therefore a need for compounds with
exact or similar structure to those glyconjugates in order to study their
function and eventually to interfere in some biological pathways.
Being involved in the use of very
available carbohydrates for chemistry, we have been interested in the
preparation of some sucrose derivatives close to natural compounds such as
derivatives similar to gallotannins found in some Chinese rhubarbs having
antioxidant properties or some glycolipids analogues of Cord factors with
lamellar thermotropic behaviour. The synthesis of such compounds as well as some
of their properties will be described, with a focus on how to overcome the
inherent difficulties of regioselectivity when starting from unprotected
substrates without multiplying the
protection-deprotection steps.
Also,
we will present some recent progress in the preparation of some neoglycoconjugates based on the use of CMGLs
(carboxymethyl glycoside lactones) which are bicyclic lactones easily
obtained either by degradation of available disaccharides
or by construction from monosaccharides. Compounds in the families of pseudo-disaccharides, pseudo-glycopeptides and
pseudo-glycolipids will be described.
For recent relevant work of our group, see: Gallic esters of sucrose as efficient radical scavengers in lipid peroxidation, C. Dufour, E. Da Silva., P. Potier, Y. Queneau and O.
Dangles, J. Agric. Food. Chem.,
50, 3425-3430 (2002); A bilayer to
monolayer phase transition in liquid crystal glycolipids, V. Molinicr,
P.H.J. Kouwer, Y. Queneau, J. Fitremann, G. Mackcnzic et J. W, Goodby J. Chem. Soc., Chem. Commun,, 2860-2861 (2003); Straightforward route for anchoring a glucosyl moiety on nucleophilic species: ręaction of amines and
alcohols with carboxymethyl 3,4,6-tri-O-acetyl-a-D-glucopyranoside 2-O-lactone, J. Org. Chem., 68, 6672-6678 (2003); The chemistry of unprotected sucrose: the selectivity issue, Y. Quencau, J. Fitremann and S. Trombotto, C. R. Chimie., 7, 177-188 (2004).
|
SYNTHESIS AND ANTIBODY
RECOGNITION OF CHLAMYDIAL LIPOPOLYSACCHARIDE
Alla ZAMYATINA*a, Harald SEKLJICb,
Helmut BRADEb,
Stephen V. EVANSc, Paul KOSMAa
a Dept. of Chemistry, Univ. of Natural Resources and Applied Life
Sciences, Vienna
bMedical and Biochemical Microbiology, Research Center Borstel, Germany
c Dept. Biochemistry and Microbiology, Univ.Victoria, Canada
alla.zamyatina@boku.ac.at; paul.kosma@boku.ac.at
Chlamydiae are obligatory intracellular Gram-negative pathogens which
are responsible for a variety of acute and chronic diseases in animals and
humans, such as urogenital infections and trachoma [1]. In addition, Chl.
pneumonia infections may be associated with atherosclerosis. Although
chlamydial LPS is at least ~10 times
less active than enterobacterial endotoxins, its role in local chronic
infections and inflammatory processes needs to be clarified [2]. Based on the
structural data on C. trachomatis serotype L2 LPS [3], chlamydial tetraacyl
Lipid A and pentaacyl Lipid A has
been synthesized and was fully characterized.
In addition, synthesis of neoglycoconjugates containing
chlamydia-specific and cross-reactive Kdo-ligands allowed for a detailed
characterization of the binding of monoclonal antibodies to these bacterial
epitopes using serology, NMR-methodology and crystallography [4].
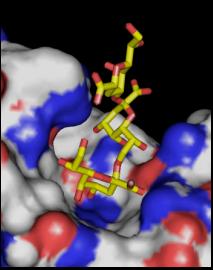
Binding of a Kdo trisaccharide to mAb S-25 Pentaacyl chlamydial Lipid A
Acknowledgments: Financial support
by FWF (P13843-CHE and P 17407)
References:
[1] Moulder, J. W. Microbiol. Rev. 1991,
55,
143-190.
[2] Heine, H.; Müller-Loennies, S.; Brade,
L.; Lindner, B.; Brade. H. Eur. J. Biochem. 2003, 270,
440-450.
[3] Zamyatina,
A., Sekljic, H., Brade, H., Kosma, P. Carbohydr.
Res. 2004, 60, 12113-12137.
[4]
Nguyen, H.P., Seto, N.O.L., MacKenzie, C.R., Brade, L., Kosma, P., Brade, H.,
Evans, S.V., Nature
Struct. Biol. 2003, 10, 1019-1025. Nature Struct.
Biol. 2003, 10, 1019-1025.
|
THIOGLYCURONIDES: SYNTHESIS
AND APPLICATION IN THE ASSEMBLY OF ACIDIC OLIGOSACCHARIDES
Leendert J. VAN DEN BOS, Herman S. OVERKLEEFT, Gijsbert A. VAN
DER MAREL
Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300
RA Leiden, The Netherlands
e-mail: l.j.vdbos@chem.leidenuniv.nl
Uronic
acids are present in a wide array of biologically relevant oligosaccharides,
polysaccharides and glycoconjugates.
Hence, flexible and straightforward synthesis routes towards these important
molecules should have major impact on research in glycobiology. Although it is
well established that thioglycosides are versatile synthons en route towards
such carbohydrate motives, approaches in which thioglycuronic acids are
employed are scarce. This can be explained by the lack of efficient synthetic
protocols for the preparation of suitably protected thioglycuronides. In
addition, thioglycuronic acids have been shown to be rather poor glycosyl
donors, generally requiring the presence of activating protecting groups.
The
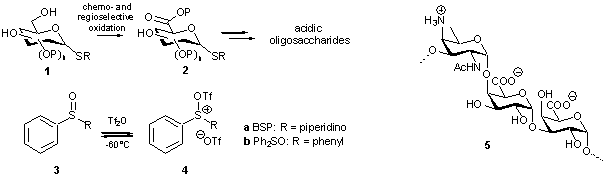
potency of the recently developed novel sulfonium based activator systems 4a and 4b encouraged us to implement the highly unreactive
thioglycuronides (2) in our recently
developed glycosylation sequence
to effectively provide acidic oligosaccharides. This evidently called for an
efficient mode of synthesis to access a wide variety of thioglycuronic acid
synthons. We here present the 2,2,6,6-tetramethyl-1-piperidinyloxyl
(TEMPO)/[bis(acetoxy)-iodo]benzene (BAIB) mediated chemo- and regioselective oxidation
of readily available partially protected thioglycosides as a powerful means to
obtain the corresponding thioglycuronic acids.
After esterification of the carboxylate functions, these partially protected
thioglycuronides 2 can be
incorporated in the synthesis towards acidic oligosaccharides (e.g.
trisaccharide 5).
FUNCTIONALIZATION OF THE HOMOALLYLIC BRIDGE IN HIGHER
SUGAR PRECURSORS
Sławomir JAROSZ, Katarzyna SZEWCZYK, Anna
GAWEŁ
Institute of Organic Chemistry, Polish Academy of
Sciences, Kasprzaka 44/52, 01-224 Warszawa, POLAND; korana@icho.edu.pl
Recently we proposed a convenient
method of the synthesis of higher sugar homoallylic alcohols (2) from allyltin derivatives 1.1 Suitable
functionalization of the C-2 and C-3 carbon atoms might provide the
2-hydroxy-3-deoxy derivative 4. This
structural unit occurs in e.g.
11-carbon atom antibiotic sugar – tunicamine.2
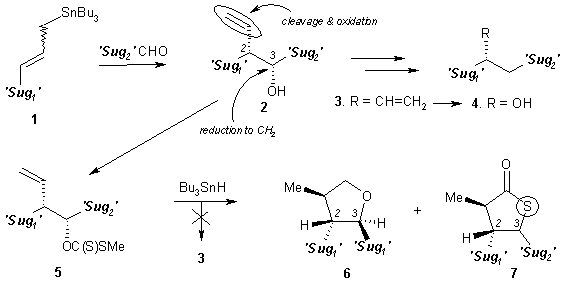
We faced a big problem in conversion
of compound 2 into a deoxy higher
sugar precursor 3. The Barton-McCombie reduction3
of the hydroxyl group in 5 did not
afford the expected compound 3 but,
provided two other products 6 and 7.4 Mechanism of these
transformations will be discussed.
1.
Jarosz, S.;
Szewczyk, K.; Luboradzki, R.; Gaweł, A. Tetrahedron:
Assymetry, 2004, 15,
1719.
2.
Takatsuki, A.;
Arima, G.; Tamura, J. J. Antibiot., 1971, 24, 215.
3.
review: Crich, D.;
Quintero, L. Chem. Rev., 1989, 89, 1413.
4.
Jarosz, S.;
Szewczyk, K.; Gaweł, A.; Gomez, A.M.; Lopez, J.C. Polish J. Chem., 2005, 79, 231.
SYNTHESIS OF STRUCTURAL ANALOGS OF NATURAL PRODUCTS
Pál HERCZEGH
Department of Pharmaceutical
Chemistry, University of Debrecen and
Research Group for Chemistry of
Antibiotics of the Hungarian Academy of Sciences
H-4010 Debrecen, Hungary
The following topics will be discussed:
- Swainsonin and castanospermin analogs
Polyhydroxyindolizidines and
quinolizidines have been prepared with the use of hetero-Diels-Alder and
1,3-dipolar cycloaddition reactions of sugar derivatives.
- Cycloadditions of nitrilimines
Stereochemistry of inter- and
intramolecular cycloaddition reactions of sugar derived nitrilimines have been
studied.
- Synthesis of sialyl Lewis X
oligosaccharide analogs
Sulfonyl Lewis X derivatives were
synthesized.
- Preparation of conagenin antibiotic
analogs
Diastereoisomers of the immunostimulant antibiotic conagenin was
performed starting from simple sugars.
- A new synthesis of 2-deoxyamino sugar
glycosides.
Some simple 2-deoxyamino sugar glycosides
have been prepared using sulfonylnitrene additions of glycals.
- A glycan synthesis by sugar
polymerisation
The poly-Ferrier reaction of a glucal
derivative led to a mixture of unsaturated glycans.
TRIMETHYLENE DITHIOACETALS OF CARBOHYDRATES: NEW DEVELOPMENTS
Hartmut REDLICH
Organisch-Chemisches Institut der Westfälischen
Wilhelms-Universität,
Corrensstraße 40, 48149 Münster, Germany
The dithian function is a powerful
instrument in organic synthesis, due to its value for the concept of 'Umpolung'. Sugar dithians are easily available
from all basic sugars. They
have found various applications in natural product syntheses. This lecture will deal with
developments in applying trimethylene dithioacetals of carbohydrates in new
synthetic fields, covering:
1.
Sugar dithians in glycoside syntheses
Closing a missing link
2.
Orthoesters of sugar dithians
A surprisingly easy entrance for a
new protecting group strategy on open chain polyols
3.The
trimethylene dithioacetal of D-glucosamine
Intramolecular C-C and C-N bond formation to yield highly substituted
N-containing carbacycles or iminosugars
A CONVENIENT ROUTE TO HIGHLY OXIDIZED CARBOBICYCLES FROM SUGAR ALLYLTINS
Sławomir
JAROSZ, Marcin NOWOGRÓDZKI
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warszawa, Poland;
mnowogro@icho.edu.pl
Higly oxidized analogues of decalin
such as 1 are attractive drug candidates. Their ability to inhibite function
of glicosydases are thought to originate from carbocyclic skeleton
(carboanalogues of sugars).
Stereoselective synthesis of such
molecules is of interest in our group. We examine the “chiral pool” approach to
derivatives of 1 from simple sugars (an example from glucose).
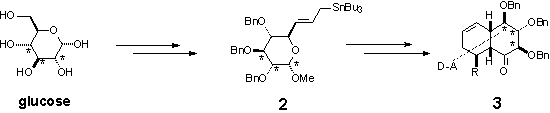
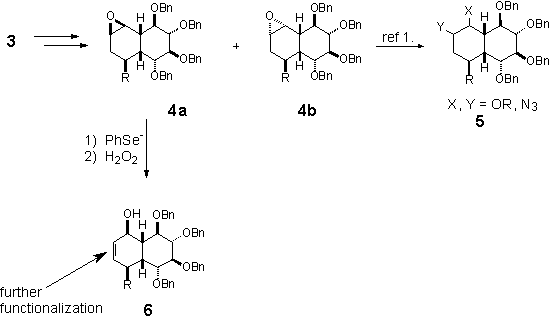
The derivative 3 is further
oxidized into epoxides 4a and 4b. Opening of the oxirane ring
with different nucleophiles led to interesting products 5.1 Opening
with selenide anion and further oxidation/elimination of selenoxide led to the
product of formal basic rearrangement of the epoxide 4a, which is
further modified towards highly substituted decalin derivatives.
- Jarosz,
S.; Skóra, S. Tetrahedron: Asymmetry,
2000, 11, 1433 – 1448
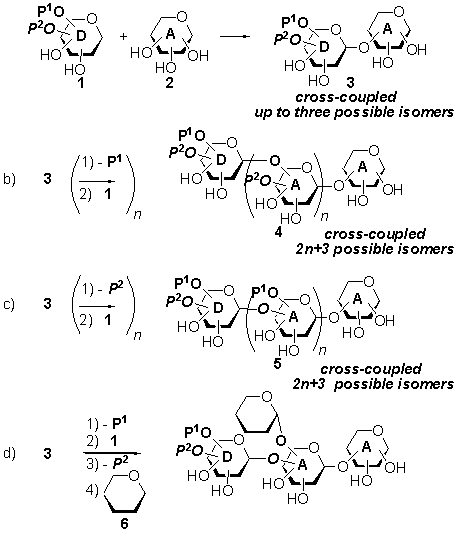
ITERATIVE, ORTHOGONAL STRATEGY FOR OLIGOSACCHARIDE SYNTHESIS BASED ON
THE REGIOSELECTIVE GLYCOSYLATION OF TRIOL ACCEPTORS WITH PARTIALLY UNPROTECTED
N-PENTENYL-ORTHOESTERS
A. M. GÓMEZ, A. AGOCS, C. URIEL, B.
FRASER-REID, J. Cristóbal LÓPEZ
Instituto de Química Orgánica General
(CSIC, Madrid, SPAIN) and a Natural Products and Glycotechnology Research Institute Inc.,(NPG),Durham, North Carolina, USA
We have studied an iterative
protocol based on the regioselective glycosyl coupling of D-mannose triols
(e.g. 2) with partially unprotected n-pentenyl orthoester
glycosyl donors (e.g. 1) (a, Scheme) which, permits the synthesis
of linear and branched oligosaccharides with minimum protecting groups tampering. In
this strategy, the glycosyl donor possesses two orthogonal protecting groups
which can be selectively manipulated thus paving the way for regioselective
glycosidation strategies (b, c or d, Scheme) leading to linear (b or c, Scheme)
or branched (d, Scheme) oligosaccharides.
LEWIS ACID CATALYZED
DIASTEREOSELECTIVE ALLYLATION OF CHIRAL ACTIVATED KETONES
Tomasz
BAŁAKIER,a Janusz JURCZAKa,b
a Department of Chemistry,
University of Warsaw, 02-093 Warsaw
b Institute of Organic
Chemistry, Polish Academy of Sciences, 01-224 Warsaw
jurczak@icho.edu.pl
Allylation of carbonyl compounds has
become a well established methodology for the stereoselective construction of
carbon-carbon bonds, providing an elegant synthesis of allylic alcohols.
However, due to the lower reactivity of ketones, it has been examined mostly
for aldehydes.1 This fact prompted us to study the diastereoselectivity
in the allylation reaction of chiral activated ketones, such as pyruvic and
phenylglyoxylic acid derivatives, leading to formation of quaternary
stereogenic centers.
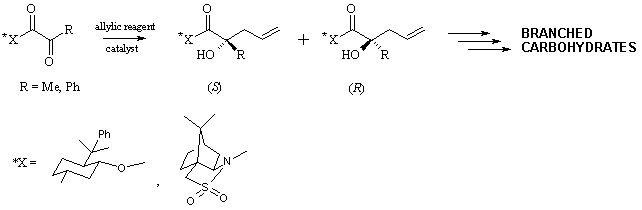
We tested variety of allylic
reagents in the reaction with chiral pyruvates and phenylglyoxylates. The
influence of Lewis acids on the reactivity and diastereoselectivity of the
reaction was also studied. The desired allylic alcohols were obtained in good
to excellent yield and diastereoselectivity.
1. Denmark, S.E.; Fu, J. Chem. Rev. 2003, 103, 2763